Department of Chemistry

- Department of Chemistry
- People
- Professor Peter O'Brien
- O'Brien Group Webpage
- Research Highlights
Peter O'Brien's Group Research Highlights
The POB group research areas focus on all aspects of novel chiral base chemistry. This includes the synthesis of new chiral bases, the development of methodology and applications in the synthesis of biologically interesting natural products. We are especially keen to develop simple, robust and reliable synthetic methods.
We are extremely grateful to all of the following for funding the research we have undertaken over the last few years:
EPSRC, BBSRC, The University of York, The Leverhulme Trust, The Royal Society, The Nuffield Foundation, EU, Merck, Celtic Catalysts, Eli Lilly, Astra-Zeneca, GlaxoSmithKline, Hoffman-la-Roche, Lancaster Synthesis (as was), Parke-Davis (as was), Pfizer, Rhone-Poulenc-Rorer (as was) and SmithKline Beecham (as was).
Some of our published research highlights together with leading references are provided in the following sections.
- (+)-Sparteine surrogate for asymmetric synthesis
- Catalytic asymmetric deprotonation
- Catalytic asymmetric synthesis of P-stereogenic phosphines
- Lithiated aziridines
- Aziridinium ion chemistry
- Chiral base-mediated epoxide rearrangement reactions
- New routes for the total synthesis of natural products
- Miscellaneous stereoselective methodology
(+)-Sparteine surrogate for asymmetric synthesis
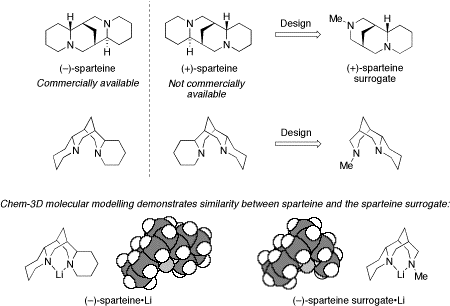
We have introduced an easy-to-synthesise (three steps) diamine ((+)-sparteine surrogate) that behaves as the enantiomer of (–)-sparteine, a naturally occurring alkaloid extracted from Scotch broom plants. (–)-Sparteine is a very useful and widely used chiral ligand for asymmetric synthesis but there is a fundamental limitation of using it in synthesis: (–)-sparteine is only commercially available in one enantiomeric form. Some time ago, we set about addressing this limitation. Our approach uses the structure of (–)-sparteine as a guide: on inspection, we reasoned that the (+)-sparteine surrogate, which lacks one of the rings and chiral centres of (+)-sparteine, would be a good (+)-sparteine mimic.
Our key contribution was the realisation that the naturally occurring alkaloid, (–)-cytisine, is equipped with the required bispidine framework and the correct absolute stereochemistry needed for our proposed (+)-sparteine mimic. The synthesis of the (+)-sparteine surrogate is outlined below. First of all, (–)-cytisine was obtained by extraction from Laburnum anagyroides seeds: 3.5-3.9 g (approx. 1.5% mass yield) of (–)-cytisine can be isolated from 200 g of seeds. Then, protection of the amine as a methyl carbamate was followed by hydrogenation and lithium aluminium hydride reduction to give multi-gram quantities of the diamine.
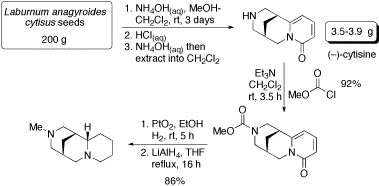
This synthesis of the (+)-sparteine surrogate has been published in Organic Syntheses: Org. Synth., 2006, 83, 141.
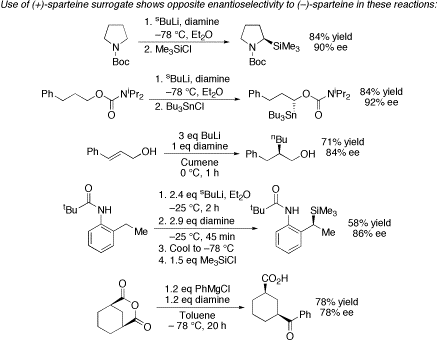
To illustrate that the (+)-sparteine surrogate did indeed behave as the “enantiomer of (–)-sparteine”, we carried out a number of “test” reactions. Several examples are shown below. In each case, use of the (+)-sparteine surrogate led to products with the opposite sense of asymmetric induction compared to that obtained with (–)-sparteine, but with similar degrees of enantioselectivity.
Other research groups have used our methodology to prepare the (+)-sparteine surrogate and then used it in their own chemistry:
N. C. Kann: Eur J Org Chem, 2004, 1894; Tetrahedron: Asymmetry, 2004, 15, 3531; Synlett, 2006, 3389; J Comb Chem, 2007, 9, 477.
J. A. Wilkinson: Tetrahedron: Asymmetry, 2004, 15, 3011; Tetrahedron, 2006, 62, 1833.
T. Fukuyama, Org Lett, 2005, 7, 4337.
V. K. Aggarwal, Angew Chem Int Ed, 2007, 46, 7491.
The usefulness of the (+)-sparteine surrogate has been critically reviewed: R. Iyengar and V. Gracias, Chemtracts–Organic Chemistry, 2004, 17, 92.
In order to probe the alkyllithium-diamine reactivity further, we have initiated collaborations in three areas:
(i) Computational-based approach to organolithium chemistry – collaboration with Professor K. B. Wiberg (Yale University, New Haven, USA) and Professor W. F. Bailey (University of Connecticut, Connecticut, USA);
(ii) X-ray structures of organolithium complexes – collaboration with Dr C. Strohmann (TU Dortmund, Germany)
(iii) Solution structures of organolithium complexes – collaboration with Professor G. Hilmersson (Göteborg. Sweden)
A Chem. Commun. Feature Article on the (+)-sparteine surrogate has been published:
Basic Instinct: Design, Synthesis and Evaluation of (+)-Sparteine Surrogates for Asymmetric Synthesis, P. O’Brien, Chem. Commun., 2008, 655-667.
Leading references on the (+)-sparteine surrogate for asymmetric synthesis:
Angew Chem Int Ed, 2008, 47, 6367; Org Lett, 2008, 10, 1409; J Org Chem, 2008, 73, 6452; Org Biomol Chem, 2006, 4, 1376; Chem Commun, 2006, 2607; Org Synth, 2006, 83, 141; J Am Chem Soc 2004, 126, 15480; Organometallics, 2004, 23, 5389; J Org Chem, 2004, 69, 5789; Org Biomol Chem, 2003, 1, 3977; J Am Chem Soc, 2002, 124, 11870; Chem Commun, 2001, 1202; J Chem Soc, Perkin Trans 1, 1999, 3623.
Catalytic asymmetric deprotonation
Catalytic asymmetric synthesis using organolithium reagents and sub-stoichiometric amounts of chiral diamines such as (–)-sparteine is a challenging research area. However, our group has had recent, unprecedented success in developing some catalytic asymmetric deprotonation reactions.
We have successfully developed a two-ligand approach for a catalytic asymmetric Beak-style deprotonation of N-Boc pyrrolidine. A comparison between the stoichiometric (–)-sparteine result and a reaction that utilises just 0.2 equiv. (–)-sparteine is shown below. The catalysis example is only successful if a second, sterically hindered bispidine is included in the reaction mixture. We believe that the bispidine allows efficient recycling of the (–)-sparteine via a ligand exchange process. This two-ligand approach is also successful for the catalytic asymmetric deprotonation of O-alkyl carbamates, as introduced by Hoppe.
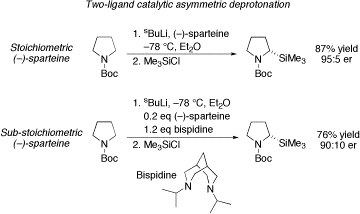
In contrast, Snieckus-style asymmetric lithiation-trapping of a ferrocene amide is successful using one-ligand catalysis. Using only 0.4 equiv. (–)-sparteine, a similar yield and enantioselectivity to the stoichiometric example was obtained.

We have also discovered that better yields and/or higher enantioselectivity are obtained using the (+)-sparteine surrogate in the catalytic asymmetric deprotonation reactions.
Leading references on catalytic asymmetric deprotonation:
J Org Chem, 2008, 73, 6452; J Am Chem Soc, 2006, 128, 9336; Chem Commun, 2006, 2607; Synthesis, 2006, 2233; J Am Chem Soc, 2005, 127, 16378.
Catalytic asymmetric synthesis of P-stereogenic phosphines
We have recently embarked on a programme of research aimed at developing the catalytic asymmetric synthesis of P-stereogenic phosphines as these are one of the most important classes of chiral ligands for transition metal-catalysed asymmetric processes. The strategy we are employing is one-ligand catalytic asymmetric deprotonation of activated phosphine derivatives (eg phosphine boranes), a reaction originally introduced by Evans. Crucially, use of the (+)-sparteine surrogate allows convenient access to the opposite enantiomeric series to that obtained using (–)-sparteine.
Selected results using the (+)-sparteine surrogate are shown below, including applications in the lithiation-dimerisation process which is a convenient way of generating protected bisphosphine ligands.
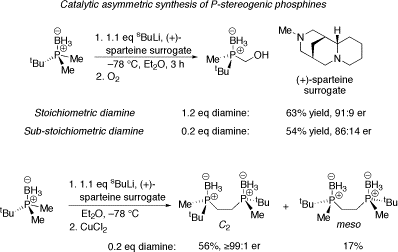
Leading reference on catalytic asymmetric synthesis of P-stereogenic phosphines:
Chem. Commun. 2008, 3750; J Am Chem Soc, 2006, 128, 9336.
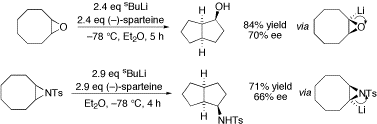
Lithiated aziridines
We have reported a number of key observations in the area of lithiated N-tosyl aziridines, generated by direct deprotonation. An important finding (shown below) was that cyclooctene oxide and its aziridine analogue are lithiated with opposite senses of induction using s-butyllithium and (–)-sparteine. The enantioselectivity was, however, similar in each case.
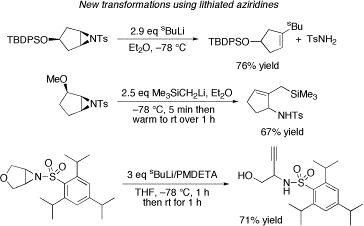
In addition, we are the first group to report examples of the three new transformations of lithiated aziridines shown below. Thus, upon treatment with organolithium reagents, aziridines have been transformed into substituted alkenes, substituted allylic sulfonamides and alkynyl amino alcohols. All of these reactions are believed to proceed via lithiated aziridines and their scope and usefulness in synthesis has been fully explored. In particular, the substituted allylic sulfonamide-forming reaction has been used in the development of a new route to azaspirocycles.
Leading references on lithiated aziridine chemistry:
Org Biomol Chem, 2009, 1, 335; Synlett, 2008, 237; J Org Chem, 2008, 73, 7852; Org Biomol Chem, 2008, 6, 4299; Org Lett, 2006, 8, 5145; Chem Commun, 2005, 5696; Org Lett, 2004, 6, 4817; Tetrahedron, 2003, 59, 9779; Tetrahedron Lett, 2003, 44, 6613; Org Lett, 2002, 4, 1923.
Aziridinium ion chemistry
We have developed a convenient one-pot method for the synthesis of chiral diamines starting from styrene oxide in which the regio- and stereospecific ring opening of an aziridinium ion (R = Ph) is the key step. A detailed study on the factors controlling the regioselectivity of aziridinium ion ring opening was carried out: attack of MeNH2 occurs at the more hindered position of when R = Ph (electronic control) and at the least hindered position when R ≠ Ph (steric control). Extension of our approach allowed a N,N-dialkylated norephedrine to be converted into a novel diamine in high yield via regioselective benzylic opening of an aziridinium ion.
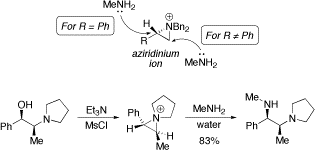
Our aziridinium ring-opening methodology has been successfully scaled up to the multi-kilo scale by process chemists at Amgen: Org. Proc. Res. Dev., 2007, 11, 215.
Leading references on aziridinium ion chemistry:
J Org Chem, 2002, 67, 304; Tetrahedron Lett, 2002, 43, 7333; Synthesis, 2001, 693; Tetrahedron: Asymmetry, 1999, 10, 4175; J Chem Soc, Perkin Trans 1, 1998, 1483; Tetrahedron Lett, 1996, 37, 5619.
Chiral base-mediated epoxide rearrangement reactions
The use of chiral bases (typically derived from diamines) for the enantioselective rearrangement of epoxides to allylic alcohols has been an ongoing research interest of ours and we have made two useful contributions in this area: we extended the scope of epoxide rearrangements to highly functionalised epoxides and we developed a highly enantioselective norephedrine-derived chiral base. As examples, all of the allylic alcohols shown below have been prepared as useful building blocks for synthesis.
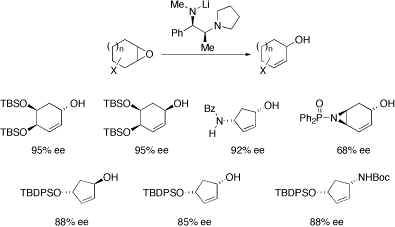
The epoxide rearrangement methodology has been successfully scaled up to the multi-kilo scale by process chemists at Eli Lilly: Org. Proc. Res. Dev., 2007, 11, 546.
Leading references on chiral base-mediated epoxide rearrangement reactions:
Tetrahedron Lett, 2005, 46, 8315; Tetrahedron Lett, 2004, 45, 9053; Org Biomol Chem, 2003, 1, 523; Tetrahedron, 2002, 58, 4643; Tetrahedron, 2000, 56, 9633; Chem Commun, 2000, 1521; Tetrahedron Lett, 1999, 40, 8423; J Chem. Soc, Perkin Trans 1, 1998, 2435.
Keynote review on chiral lithium amide bases: P. O’Brien, J Chem Soc, Perkin Trans 1 1998, 1439.
Tetrahedron Symposium-in-Print: Guest editor P. O’Brien, “Recent Developments in Chiral Lithium Amide Base Chemistry”. Tetrahedron, 2002, 58, 4567-4733.
New routes for the total synthesis of natural products
We are also interested in utilising chiral base methodology and related methodology in natural product synthesis. Some of the target molecules that have been or continue to be of interest are shown below.
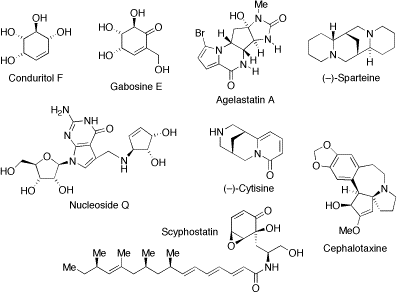
(±)-Cytisine. (–)-Cytisine is a naturally occurring lupin alkaloid that exhibits partial agonist activity at neuronal nicotinic acetylcholine receptors with specificity for the α4ß2 sub-type. Currently, there is much interest in the development of “cytisine-like” nicotinic agonists for the treatment of various CNS disorders and for assisting smoking cessation. We have developed a 6-step synthesis of (±)-cytisine (19% overall yield). Key features of this new strategy (shown below) include: (i) initial construction of the bispidine core, (ii) lithiation-transmetallation-allylation of an N-Boc bispidine, (iii) ring-closing metathesis and (iv) a Pd/C-mediated dihydropyridone oxidation-N-debenzylation process.
Org Lett, 2005, 7, 4459; Tetrahedron, 2007, 63, 1885
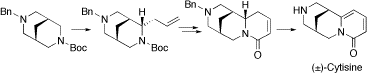
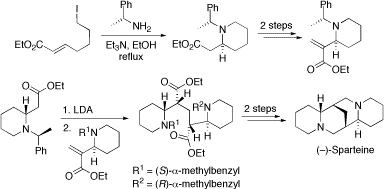
(–)-Sparteine. Our group has a long-standing interest in chiral ligands based on (–)-sparteine (see separate section). We have completed the second asymmetric synthesis of sparteine using a new strategy that could prove useful for the synthesis of other members of the lupin alkaloids. Our approach (outlined below) utilises some new methodology for the synthesis of cyclic ß-amino acids. The key step was a connective Michael reaction which sets up all four of the stereogenic centres in the natural product. Removal of the N-protecting groups and amide reduction then gave (–)-sparteine.
Chem Commun, 2004, 1830; Org Biomol Chem, 2007, 5, 3614
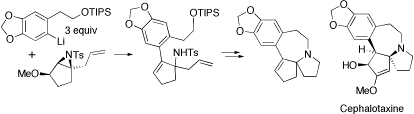
Core of Cephalotaxine. Naturally occurring esters derived from cephalotaxine (eg deoxyharringtonine) show pronounced anti-leukemic activity and have thus been the subject of several recent synthetic studies. We have developed a new synthesis of the tetracyclic core of cephalotaxine. The key step is the reaction of a functionalized aryllithium with a lithiated aziridine to give an allylic sulfonamide which was readily converted into the required azaspirocycle.
Org Lett, 2006, 8, 5145
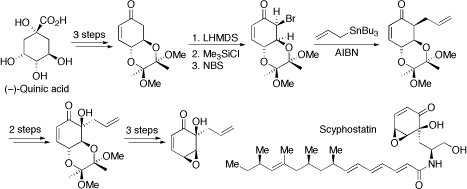
Core of Scyphostatin. Scyphostatin is a cyclohexenone-based naturally occurring inhibitor of neutral sphingomyelinase and has attracted significant synthetic interest in recent times. Starting from (–)-quinic acid, we have developed a convenient and highly stereoselective route to the cyclohexenone core of scyphostatin. The essence of our approach is summarised below and the key building block was an enone that was prepared in three steps from (–)-quinic acid. Allylation of the enone was only possible via radical-mediated allylation of the intermediate α-bromo compound. Then, stereoselective hydroxylation was carried out and the protected trans-1,2-diol functionality was unmasked to give an epoxy-enone, a suitably functionalised version of the cyclohexenone core of scyphostatin.
Org Lett, 2003, 5, 1943
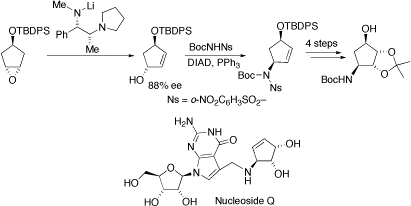
Formal synthesis of the cyclopentene core of nucleoside Q. Nucleoside Q, widely distributed in tRNAs of plants and animals, is of interest as its deficiency is related to tumour growth. We have used our chiral base methodology to prepare an allylic alcohol in 88% ee. The amino functionality was introduced via a Mitsunobu reaction and subsequent manipulations generated a known precursor to a fully protected version of the cyclopentene core of nuceoside Q.
Tetrahedron Lett, 2004, 45, 9053
Miscellaneous stereoselective methodology
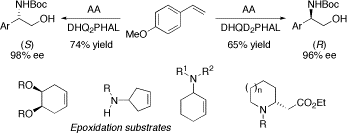
We have had a passing interest in asymmetric aminohydroxylation (AA) of alkenes. Indeed, we were the first group to report that asymmetric aminohydroxylations of styrenes with DHQ2PHAL give higher yields (but similar % ees) compared to those with DHQD2PHAL due to a better regioselectivity. We have also made important contributions in the stereoselective epoxidation of cyclic alkenes and we have developed two new routes to cyclic ß-amino acids.
Leading references:
AA reactions: J Chem Soc, Perkin Trans 1, 1998, 2519; Angew Chem, Int Ed Engl, 1999, 38, 326.
Epoxidations: Org Lett, 2003, 5, 4955; Tetrahedron Lett, 1999, 40, 391; Tetrahedron Lett, 1999, 40, 387; Tetrahedron, 2000, 56, 9633.
Cyclic ß-amino acids: Synlett, 2000, 1336; Chem Commun, 2004, 1830; Org Biomol Chem, 2007, 5, 3614.